Getting in front of pharma: Automated public discovery of drug candidates

A couple weeks ago Sean and I were fortunate enough to participate in another edition of Rhizome’s 7x7, this time in Beijing in collaboration with the Chinese Central Academy of Fine Arts (CAFA). I was very excited to collaborate with Sean again after our first collaboration in New York at the New Museum, and to have the chance to try something different together.
We thought about revisiting our previous project, cell.farm, which was a proposal for a cryptocurrency/distributed computing system for which the proof-of-work protocol involved computing simulation updates for an atomic-level model of a human cell (though our proposal initially suggested simulating a ribosome). Such detailed simulation of biological processes would be a boon for medical research, but simulating even the simplest cell at that resolution is so computationally demanding that it’s infeasible even for the world’s best supercomputers. But the aggregate computing power of the Bitcoin network is orders of magnitudes larger than any supercomputer, and might be able to run such a model in a reasonable amount of time. By adopting that model for in silico cells, a crucial part of medical research is essentially collectivized, and as part of our design, so too are the results of that research. The project bears similarity to Folding@Home and its crypto-based derivatives (e.g. FoldingCoin), but as far as I know none of these projects explicitly distribute ownership of the research that results from the network. There were also some design details that we didn’t have time to hash out, and we left open a big question of computational verifiability: given a simulation update from a node, how can you be certain that they actually computed that value rather than returned some random value? (Golem has this problem too, the difficulty of which is discussed a bit here).
This time around, rather than a project about medical research abstractly, we focused specifically on the pharmaceutical industry, the 1.1 trillion dollar business lying at the nexus of intellectual property law, predatory business practices, and the devaluing of human life.
The pharmaceutical industry
(For background I’m going to lean heavily on the “Pill of Sale” episode of the Ashes Ashes podcast which goes into more detail about the pharmaceutical industry – definitely worth a listen.)
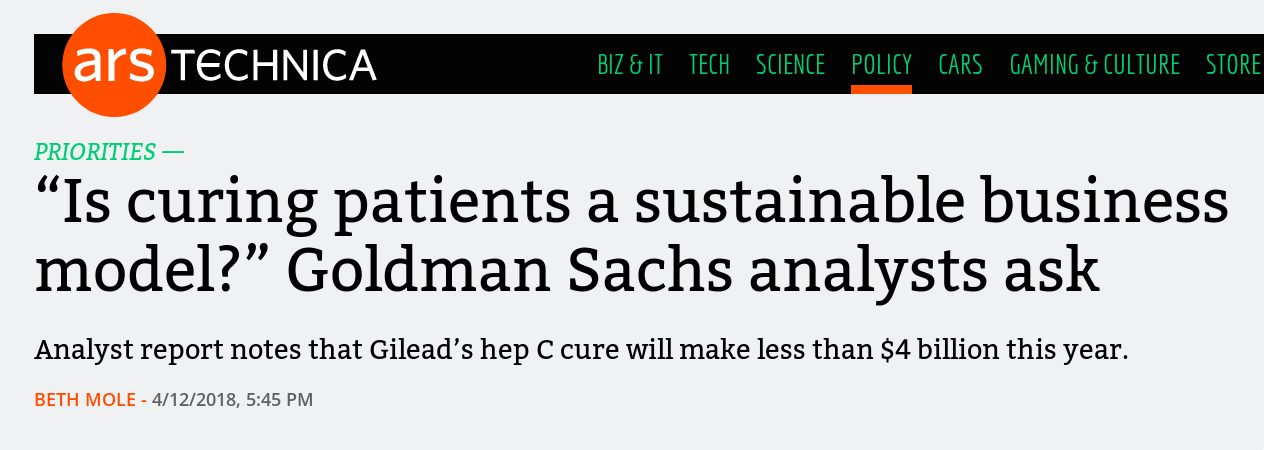
Most Americans are familiar with exorbitantly-priced drugs – if not directly than via one of the many horrifying stories of people crowdfunding their continued existence or flying elsewhere to access more reasonable prices. A hepatitis C cure from Gilead, Solvadi, costs $84,000 for a 12-week course and is the subject of a recent Goldman Sachs report. The report describes cures as effective as Solvadi (up to 97%) as bad for business since you cure yourself out of a market. Even something as common insulin can cost a significant portion of income – to the point where people die from needing to ration it.
This hostile environment is thinly justified with rhetoric around drug development costs and enforced through the patent law system, all under the implicit, sometimes explicit, assumption that it is necessary for drug companies to make a profit on their drugs. Patents provide exclusive rights for a company to sell a particular drug; this temporary monopoly essentially gives them carte blanche to set whatever price they want so that they recoup the drug development costs, so the story goes. These patents last 20 years and can basically be extended by “exclusivity” periods which add up to another 7 years. A drug may take 10-15 years to develop, leaving a window of at least 5 years of exclusive rights to produce and sell it. “Orphan drugs”, drugs that treat rare conditions, may have longer monopolies to compensate for the smaller market size. After this period generics are permitted to enter the market, which drives the cost down, but there are all sorts of tricks available that can prolong this protection period even further, a practice called “evergreening”. For example, slightly modifying how the drug is delivered (e.g. by tablet or capsule) can be enough for it to essentially be re-patented.
(It’s worth noting that prices can be high even for generics. For example, epinephrine – commonly known as an EpiPen, essential for severe allergic reactions – can be bought for about 0.10-0.95USD outside the US, whereas generics in the US can cost about $70.)
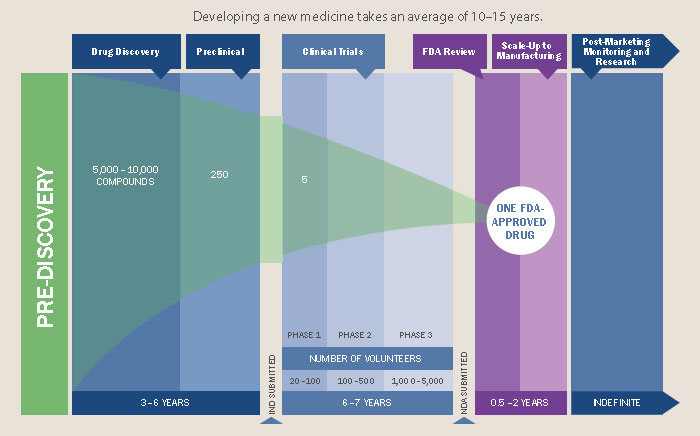
Drug development is expensive, averaging at over $2.5 billion per drug, and that’s only counting for those that gain FDA approval. However, these exclusivity rights are not merely used to recapture R&D costs, as is often said, but instead to flagrantly gouge prices such that the pharmaceutical industry is tied with banking for the largest profit margins of any industry (as high as 43% in the case of Pfizer).
The narrative around high drug development costs also takes for granted that pharmaceutical companies are the ones bearing all of these costs. A considerable amount of the basic research that is foundational to drug development is funded publicly; the linked study found that public funding contributed to every drug that received FDA approval from 2010-2016. The amount of funding is estimated to be over $100 billion.
It used to be that inventions resulting from federal funding remained under federal ownership, but the 1980 Bayh–Dole Act offered businesses and other institutions the option to claim private ownership. The result is the public “paying twice” for these drugs. The Act does preserve “march-in rights” for the government, allowing the government to circumvent the patent and assign licenses independently if the invention is not made “available to the public on reasonable terms”, but as of now these rights have never been exercised. In 2016 there was an unsuccessful attempt to use these march-in rights to lower the price of a prostate cancer drug called Xtandi, priced at $129,000/year.
All of this isn’t to say that the work of the pharmaceutical industry isn’t valuable; drugs are a necessary part of so many peoples’ lives. I recently started using sumatriptan to deal with debilitating migraines, and am hugely grateful it exists (and is not ridiculously expensive). It’s because pharmaceuticals are such a critical part to life that their development and distribution should not be dictated the values that currently shape it.
One particularly egregious example of this mess is the nightmare scenario of Purdue Pharmaceuticals, owned by the Sackler family (who are also prolific patrons of the arts), producers of OxyContin (accounting for over 80% of their sales last year), basically responsible for the ongoing opioid crisis (affecting at least 2.1 million Americans directly, and many more collaterally), and recently granted a patent for a drug that treats opioid addiction. The patented treatment is a small modification of an existing generic.
The day before our 7x7 presentation a story broke in the Guardian: “Sackler family members face mass litigation and criminal investigations over opioids crisis”.
Computational drug discovery
One reason drug development is so difficult is that the space of possible drug compounds is extremely large, estimated to be between 1060 and 1063 compounds. For comparison, there are an estimated 1022 to 1024 stars in the entire universe, and according to this estimate about 1049-1050 atoms making up our entire world.
Drug development is in large part a search problem, looking to find useful compounds within this massive space. A brute-force search is impossible; even if it took only a couple seconds to examine each possible compound you’d see several deaths of our sun (a lifespan of about 10 billion years) before fully exploring that space.
More effective techniques for searching this space include slightly modifying existing drugs for different therapeutic applications (“me-too” compounds) and literally looking at plants and indigenous medical traditions for leads (this general practice is called “bioprospecting” and this particularly colonialist form is called “biopiracy”).
Of course with the proliferation of machine learning there is a big interest in searching this space computationally. Two main categories are virtual screening (looking through known compounds for ones that look promising) and molecular generation (generating completely new compounds that look promising). We focused on molecular generation for reasons described below.
A primary goal for matter.farm is to publicize this work in computational drug discovery and also help researchers use these generated compounds as potential leads for new beneficial drugs. With our system, which is also open source, independent and institutional researchers alike can access automated drug discovery technology and hopefully accelerate the drug development process.
Prior art and public discovery of drugs
(For this section we spoke with a patent lawyer who requested that we note that they are not representing us.)
One crucial criteria for a patent is that the invention must be novel; that is, the invention cannot have already been known to the public. An existing publicly-known instance of an invention is called “prior art” and can invalidate a patent claim. However, sufficient variations to an invention may qualify it as original enough to be patentable (this is the idea behind evergreening, described above).
If a drug is discovered and made public prior to a patent claim on it, it would function as prior art and make that compound un-patentable in its current form. If we were able to generate new molecules that could function as useful drugs, and make public those new molecules, then perhaps we can prevent companies from patenting them and maintaining a temporary monopoly on their distribution.
This is the second goal of matter.farm: develop an automated drug discovery system to find and publish useful drugs so they cannot be patented.
Additional efforts
Other efforts to the address problems with pharmaceutical industry can be found in initiatives like Medicare for All and the proposed Prescription Drug Price Relief Act, and the organizing happening around those. The issues with the pharmaceutical industry are just one piece of a more general hostility in American healthcare.
There is also a burgeoning DIY medicine movement which aim to build alternatives to industrialized medicine, providing autonomy, access, and reliability where those are normally withheld. For example, the artist Ryan Hammond is working on genetically modifying tobacco plants to produce estrogen and testosterone, and the Four Thieves Vinegar Collective (discussed in the Ashes Ashes “Pill of Sale” episode) provides instructions for a DIY EpiPen and a DIY lab (“MicroLab”) for synthesizing various pharmaceuticals, including Naloxone and Solvadi.
That’s it for the background of the project. The following section describes how the system works in more detail.
The project code is available here.
How it works
The complete matter.farm system involves three components:
- A molecular generation model, using a version of the graph variational autoencoder (“JTNN-VAE”) described in [2], modified to be conditional (“JTNN-CVAE”). This generates new compounds given a receptor and action, e.g. a nociceptin receptor agonist.
- An ATC code prediction model. The Anatomical Therapeutic Chemical (ATC) classification system categorizes compounds based on their therapeutic effects. We use this to estimate what a generated compound might treat.
- A retrosynthesis planner. Retrosynthesic analysis is the process of coming up with a plan to synthesis some target compound from an inventory of base compounds (e.g. compounds you can purchase directly from a supplier). This is necessary to meet the enablement requirement of prior art; that is, it’s not enough to come up with a new compound, you also need to sufficiently demonstrate how it could be synthesized.
To train these various models we relied on a number of public data sources, including PubChem, UniProtKB, STITCH, BindingDB, ChEMBL, and DrugBank.
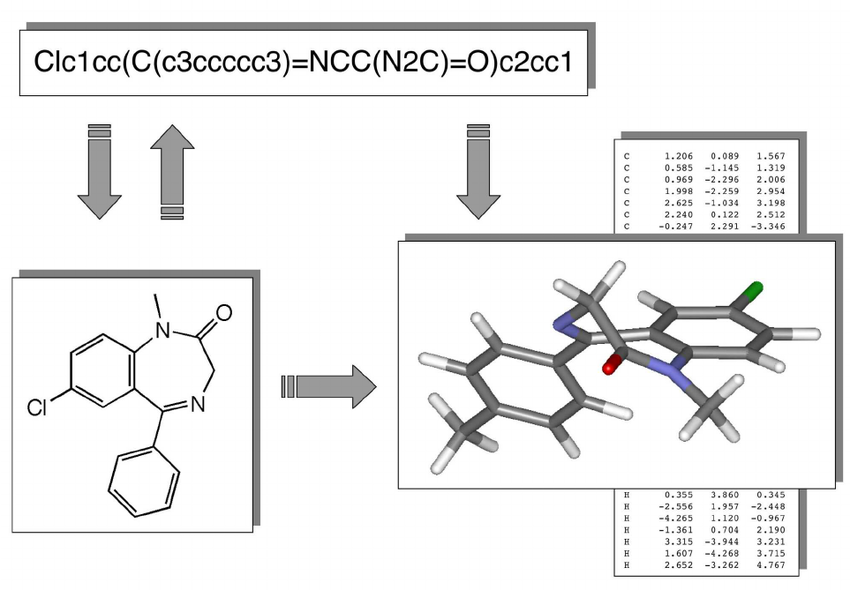
Chemical compounds can be represented in a number of ways, e.g. as a 2D structural diagram or a 3D model. One of the most portable formats is SMILES, which represents a molecule as an ASCII string, and is what we use throughout the project.
Clustering
For training the JTNN-CVAE model we needed to cluster the compounds in a meaningful way. At first we didn’t look at receptors and actions but rather tried to leverage the vast published chemical research literature (in PubMed) and USPTO patents.
For the first attempt we tried learning word2vec embeddings from PubMed article titles and abstracts, then representing documents using TF-IDF as described in the WISDM algorithm, and finally clustering using DBSCAN or OPTICS. This ended up being way too slow, memory intensive, and limited in what clustering algorithms we could try.
The second attempt involved generating a compound graph such that an edge exists between compound A and B if they appear in an article or patent together. So instead of linking compounds based on the content of the articles they’re mentioned in, they’re linked solely on the virtue of being mentioned together in an article, under the assumption that this indicates some meaningful similarity. Then we ran a label propagation community detection algorithm to identify clusters within the graph. The graph was fairly sparse however and in the end looking at the connected components seemed to be enough. There were still some limitations with speed and memory that led us to abandon that approach.
Finally we decided to cluster based on receptors compounds were known to interact with. This reduced the amount of compounds we were able to look at (since the compounds for which receptor interactions are known are much less than the total of all known compounds), but the data was richer and more explicit than co-mentions in text documents. A drug’s effects are determined by the receptor it targets and how it interacts with that receptor (does it activate it, does it block it, etc). For instance, OxyContin is a mu-type and kappa-type opioid receptor agonist (it activates them), and Naloxone (used to treat opioid overdose) is a mu-type and kappa-type opioid receptor antagonist (it blocks them).
The ChEMBL data has information about both receptors and the type of interaction (agonist, antagonist, etc), whereas BindingDB has information about only the receptors but for more compounds. So we used a two-pass approach: for the first pass, we create initial clusters based on receptor-action types and on the second-pass we augment these clusters with the BindingDB compounds, assigning them to the cluster that matches their target receptor and has the highest fingerprint similarity to the cluster’s current members. This resulted in 461 receptor-action clusters.
Molecular generation model
The JTNN-VAE model described in [2] is a variational autoencoder that handles molecular graphs. A variational autoencoder is a generative model that learns how to compress (“encode”) data in such a way that it can be reliable decompressed (“decoded”). It accomplishes this by learning an underlying probability distribution that describes the data. Once the model has learned this distribution you can sample it to generate new data that looks like the old data. This post provides an overview on variational autoencoders.
We modified the model to be conditional, allowing us to sample from the learned probability distribution conditioned on the cluster (the receptor-action) we want to generate new compounds for.
ATC code prediction model
The ATC code prediction model is a straightforward multiclass neural network. Though ATC code prediction is technically a multilabel problem (a compound may have more than one ATC code), most compounds had only one code, so we treated it as a multiclass problem. ATC codes have 5 levels, from low detail to high detail; we predict level 3 codes (“pharmacological/therapeutic/chemical subgroup”), with one class for each level 3 code. We use 2048-bit Morgan fingerprints as representations for the compounds and achieved about 80% accuracy with this naive approach – not ideal, but fine for our purposes and time constraints.
Retrosynthesis planner
Here’s where it came down to the wire. We attempted to implement the model (“3N-MCTS”) described in [7], which involves Monte Carlo Tree Search (MCTS) and three policy networks. The policy networks predict what reaction rules might apply to a given compound. The paper’s model is trained on Reaxys data, which is way too expensive for us, so we used a smaller dataset extracted from USPTO patents (from [26]). Our implementation is basically complete but we didn’t have enough time to train the models.
In an 11th-hour Hail Mary we used MIT’s ASKCOS system which got us mostly partial synthesis plans. At some point we should revisit the 3N-MCTS system to see if we can get that working.
Sampling and filtering
Once all the models were ready we sampled 100 new compounds for each of the 461 receptor-action clusters. Then we filtered down to valid compounds (with no charge) and to those not present in PubChem’s collection of 96 million compounds. We ended up whittling the set down to about 15,000 new compounds for which we then predicted ATC codes and generated synthesis plans.
Future work
Time was fairly tight for the project so we didn’t get to tune or train the system as much as we wanted to. And it would have been nice to try to experiment with more substantial modifications of the models we used. But for me this project was a wonderful learning experience and renewed an interest in chemistry. I want to spend some more time in this area, especially in materials science because of its relevance to lower-impact technologies, such as this cooling material.
References
- Botev, Viktor, Kaloyan Marinov, and Florian Schäfer. “Word importance-based similarity of documents metric (WISDM): Fast and scalable document similarity metric for analysis of scientific documents.” Proceedings of the 6th International Workshop on Mining Scientific Publications. ACM, 2017
- Jin, Wengong, Regina Barzilay, and Tommi Jaakkola. “Junction Tree Variational Autoencoder for Molecular Graph Generation.” arXiv preprint arXiv:1802.04364 (2018).
- Kusner, Matt J., Brooks Paige, and José Miguel Hernández-Lobato. “Grammar variational autoencoder.” arXiv preprint arXiv:1703.01925 (2017).
- Goh, Garrett B., Nathan O. Hodas, and Abhinav Vishnu. “Deep learning for computational chemistry.” Journal of computational chemistry 38.16 (2017): 1291-1307.
- Yang, Xiufeng, et al. “ChemTS: an efficient python library for de novo molecular generation.” Science and technology of advanced materials 18.1 (2017): 972-976.
- Liu, Yue, et al. “Materials discovery and design using machine learning.” Journal of Materiomics 3.3 (2017): 159-177.
- Segler, Marwin HS, Mike Preuss, and Mark P. Waller. “Planning chemical syntheses with deep neural networks and symbolic AI.” Nature 555.7698 (2018): 604.
- Kim, Edward, et al. “Virtual screening of inorganic materials synthesis parameters with deep learning.” npj Computational Materials 3.1 (2017): 53.
- Josse, Julie, Jérome Pagès, and François Husson. “Testing the significance of the RV coefficient.” Computational Statistics & Data Analysis 53.1 (2008): 82-91.
- Cordasco, Gennaro, and Luisa Gargano. “Community detection via semi-synchronous label propagation algorithms.” Business Applications of Social Network Analysis (BASNA), 2010 IEEE International Workshop on. IEEE, 2010.
- Community Detection in Python
- Jin, Wengong, Regina Barzilay, and Tommi Jaakkola. “Junction Tree Variational Autoencoder for Molecular Graph Generation.” arXiv preprint arXiv:1802.04364 (2018).
- What are the differences between community detection algorithms in igraph?
- Summary of community detection algorithms in igraph 0.6
- Wang, Yong-Cui, et al. “Network predicting drug’s anatomical therapeutic chemical code.” Bioinformatics 29.10 (2013): 1317-1324.
- Liu, Zhongyang, et al. “Similarity-based prediction for Anatomical Therapeutic Chemical classification of drugs by integrating multiple data sources.” Bioinformatics 31.11 (2015): 1788-1795.
- Cheng, Xiang, et al. “iATC-mHyb: a hybrid multi-label classifier for predicting the classification of anatomical therapeutic chemicals.” Oncotarget 8.35 (2017): 58494.
- Szklarczyk D, Santos A, von Mering C, Jensen LJ, Bork P, Kuhn M. STITCH 5: augmenting protein-chemical interaction networks with tissue and affinity data. Nucleic Acids Res. 2016 Jan 4;44(D1):D380-4.
- Wishart DS, Feunang YD, Guo AC, Lo EJ, Marcu A, Grant JR, Sajed T, Johnson D, Li C, Sayeeda Z, Assempour N, Iynkkaran I, Liu Y, Maciejewski A, Gale N, Wilson A, Chin L, Cummings R, Le D, Pon A, Knox C, Wilson M. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2017 Nov 8. doi: 10.1093/nar/gkx1037.
- Kim S, Thiessen PA, Bolton EE, Chen J, Fu G, Gindulyte A, Han L, He J, He S, Shoemaker BA, Wang J, Yu B, Zhang J, Bryant SH. PubChem Substance and Compound databases. Nucleic Acids Res. 2016 Jan 4; 44(D1):D1202-13. Epub 2015 Sep 22 [PubMed PMID: 26400175] doi: 10.1093/nar/gkv951.
- Gilson, Michael K., et al. “BindingDB in 2015: a public database for medicinal chemistry, computational chemistry and systems pharmacology.” Nucleic acids research 44.D1 (2015): D1045-D1053.
- The UniProt Consortium. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 45: D158-D169 (2017)
- Gaulton A, Hersey A, Nowotka M, Bento AP, Chambers J, Mendez D, Mutowo P, Atkinson F, Bellis LJ, Cibrián-Uhalte E, Davies M, Dedman N, Karlsson A, Magariños MP, Overington JP, Papadatos G, Smit I, Leach AR. (2017) ‘The ChEMBL database in 2017.’ Nucleic Acids Res., 45(D1) D945-D954.
- Papadatos, George, et al. “SureChEMBL: a large-scale, chemically annotated patent document database.” Nucleic acids research 44.D1 (2015): D1220-D1228.
- Lowe, Daniel Mark. Extraction of chemical structures and reactions from the literature. Diss. University of Cambridge, 2012.
- Lowe, Daniel (2017): Chemical reactions from US patents (1976-Sep2016). figshare. Fileset. CC0 License.
- Liu, Bowen, et al. “Retrosynthetic reaction prediction using neural sequence-to-sequence models.” ACS central science 3.10 (2017): 1103-1113.
- Klucznik, Tomasz, et al. “Efficient syntheses of diverse, medicinally relevant targets planned by computer and executed in the laboratory.” Chem 4.3 (2018): 522-532.
- Segler, Marwin HS, and Mark P. Waller. “Neural‐Symbolic Machine Learning for Retrosynthesis and Reaction Prediction.” Chemistry–A European Journal 23.25 (2017): 5966-5971.
- Law, James, et al. “Route designer: a retrosynthetic analysis tool utilizing automated retrosynthetic rule generation.” Journal of chemical information and modeling 49.3 (2009): 593-602.
- Schwaller, Philippe, et al. ““Found in Translation”: predicting outcomes of complex organic chemistry reactions using neural sequence-to-sequence models.” Chemical science 9.28 (2018): 6091-6098.
- Kipf, Thomas N., and Max Welling. “Semi-supervised classification with graph convolutional networks.” arXiv preprint arXiv:1609.02907 (2016).
- Yang, Zhilin, William W. Cohen, and Ruslan Salakhutdinov. “Revisiting semi-supervised learning with graph embeddings.” arXiv preprint arXiv:1603.08861 (2016).
- Kipf, Thomas, et al. “Neural relational inference for interacting systems.” arXiv preprint arXiv:1802.04687 (2018).
- Wei, Jennifer N., David Duvenaud, and Alán Aspuru-Guzik. “Neural networks for the prediction of organic chemistry reactions.” ACS central science 2.10 (2016): 725-732.
- Coley, Connor W., et al. “Prediction of organic reaction outcomes using machine learning.” ACS central science 3.5 (2017): 434-443.
- Gupta, Anvita. “Predicting Chemical Reaction Type and Reaction Products with Recurrent Neural Networks.”
- Plehiers, Pieter P., et al. “Automated reaction database and reaction network analysis: extraction of reaction templates using cheminformatics.” Journal of cheminformatics 10.1 (2018): 11.
- MIT’s ASKCOS (“Automated System for Knowledge-based Continuous Organic Synthesis”)